Cancer is basically a disease of cells characterized by a shift in the control mechanisms that govern cell proliferation and differentiation.
Cells that have undergone euplastic transformation usually express cell surface antigens that may be of normal fetal type, may display other signs of apparent immaturity, and may exhibit qualitative or quantitative chromosomal abnormalities, including various translocations and the appearance of amplified gene sequences.
Such cells proliferate excessively and form local tumors that can compress or invade adjacent normal structures.
A small subpopulation of cells within the tumor can be described as tumor stem cells. They retain the ability to undergo repeated cycles of proliferation as well as to migrate to distant sites in the body to colonize various organs in the process called metastasis.
Such tumor stem cells thus can express clonogenic or colony-forming capability.
Tumor stem cells often have chromosome abnormalities reflecting their genetic instability, which leads to progressive selection of sub clones that can survive more readily in the multicellular environment of the host.
Quantitative abnormalities in various metabolic pathways and cellular components accompany this neoplastic progression. The invasive and metastatic processes as well as a series of metabolic abnormalities resulting from the cancer cause illness and eventual death of the patient unless the neoplasm can be eradicated with treatment
Characteristics of cancer cells
- Clonal origin
- Immortality
- Uncontrolled proliferation
- Dedifferentiation
- Invasiveness
- Metastasis
5. Genetic instability
Cancer cell cycle
An understanding of cell- cycle kinetics is essential for the proper use of anticancer drugs
Many of the potent cytotoxic agents act at specific phases of the cycle and therefore, have activity only against cells that are in the process of division
1. S phase (DNA Synthesis )
In which synthesis of DNA occurs
The cellular content of DNA doubles
2. G1 or Pre- DNA
Synthetic phase in which cellular components required for DNA synthesis are
synthesized
Proteins and RNA are synthesized for specialized cell functions
In late G1, a burst of RNA syntheses occurs and many of the enzymes necessary for
DNA syntheses are manufactured
3. G2 or Post DNA synthesis phase
DNA synthesis ceases, protein and RNA synthesis continues and the micro tubular
precursors of the mitotic spindle are produced
4. M- phase or Mitosis
Mitosis occur G1 and G2 phase - Differentiation
In the M phase , the rates of protein and RNA synthesis diminish abruptly while the
genetic material is segregated into daughter cells.
After completion of mitosis, the cells enter either the G0 or the G1 phase
5. G0 phase
After mitosis cell may re-enter into cell- cycle or undergo in the resting phase
Cells are generally programmed to perform specialized functions

Causes of cancer
The following factors have been implicated in the etiology of cancer
- Viruses such as Epstein- Bari virus (EBV) , hepatitis – B virus (HBV) and human papilloma virus(HPV)
- Environmental and occupational hazards such as exposure to ionizing and UV radiation and exposure to various chemical carcinogens like azodyes, asbestos, benzene and polyvinyl chloride
- Diet and habits such as high fat and low- fiber diet , tobacco smoking and alcohol consumption
- Genetic factors such as inherited genetic mutations expression of oncogenes and repression of tumor suppressor gents
- Use of drugs like immunosuppressants and some alkylating agents
Cancer chemotherapy
Cancer is one of eh major causes of death. The treatment of cancers is still unsatisfactory due to creation characteristics of the cancer cells- like capacity for uncontrolled proliferation, invasiveness and metastasis
Moreover the cancer cells are our own cells unlike microbes which means that , drugs which, destroy these cells also can affect normal cells
The host defense mechanisms which help us in infections is not doing so in cancers as these cancer cells are also host cells
The cancer cells can be in a resting phase during which they are not sensitive t o anticancer drugs but can star multiplying later – resulting in recurrence .
These features have made cancer chemotherapy more difficult
Principles of cancer chemotherapy
Cancer chemotherapy is primarily used to induce and maintain a remission or response according to the following general principles
1. Drugs are used in combinations to increase efficacy inhibit the development of
resistance and minimize overlapping toxicities
2. Drugs that produce a high –fraction cell kill are preferred
3. Drugs are usually given intermittently but in high doses. This is less
immunosuppressive and generally more effective than continuous low-dose regiments
4. Toxicity is considerable and frequents blood counts and intensive clinical supports are
Essential
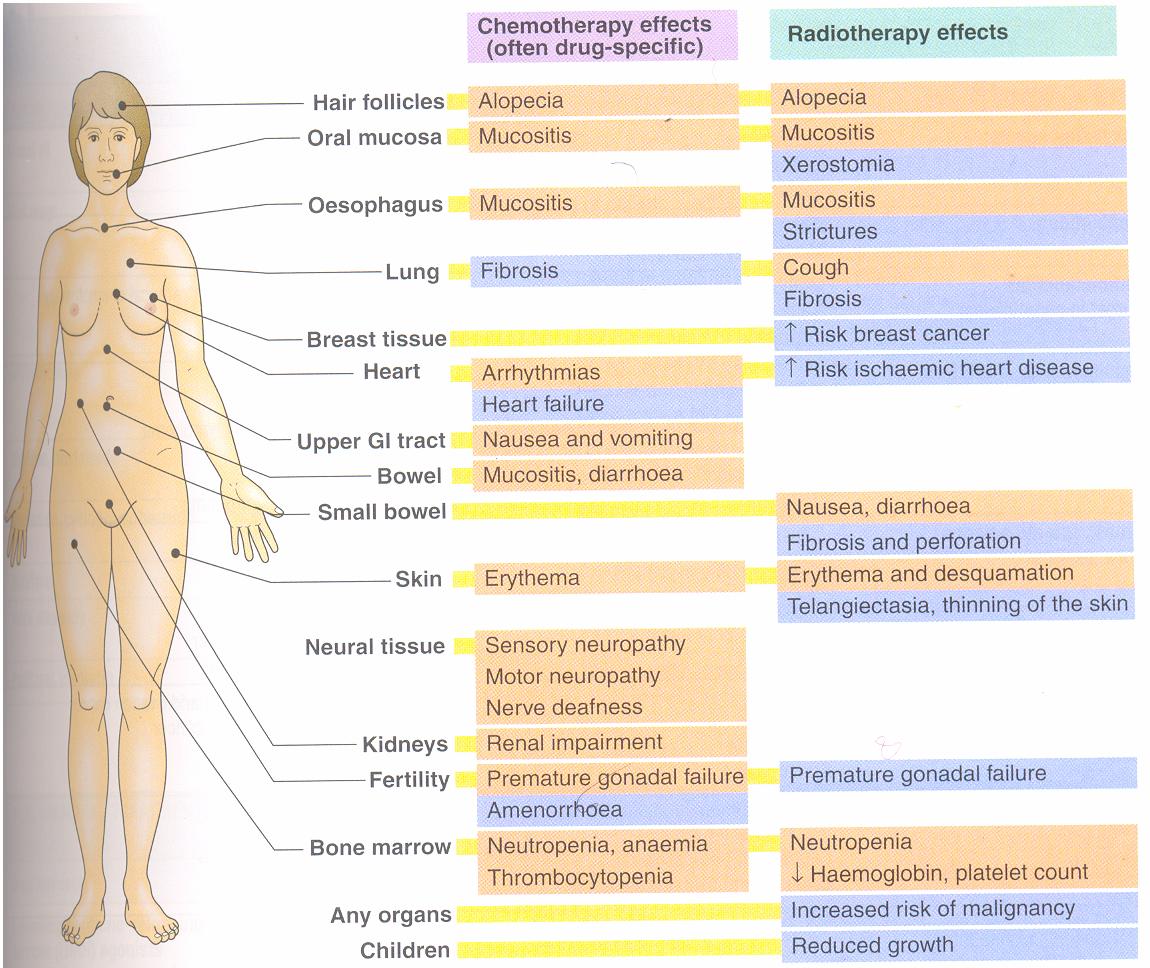
General adverse effects of anticancer drugs
Most anticancer drugs act on the rapidly multiplying cells, they are also toxic to the normal rapidly multiplying cells in the bone marrow, epithelial cells, lymphoid organs and gonads
The common adverse effects are
1. Bone marrow depression
Resulting in leucopenia, anemia, thrombocytopenia and in higher doses- a plastic
anemia, in such patients , infections and bleeding are common
2. Other effect
Alopecia , reduced spermatogenesis in men and amenorrhea in women
3. Immediate adverse effects
Nausea ,vomiting & diarrhea are very common with most
cytotoxic drugs . Prior treatment with powerful antiemetics is required
4. Psychiatric morbidity
5. Teratogeniciy
All cytotoxic drugs are teratogenic and therefore contraindicated in
pregnancy
6. carcinogenicity
Cytotoxic drugs themselves may cause secondary cancer
Eg- Leukemia’s are common after treatment of Hodgkin’s lymphoma
4. When the patient is asymptomatic with slow growing , incurable tumors , in which
case chemotherapy should be postponed until symptoms require palliation
Clinic use of cytotoxic agents
Chemotherapy is used in the following circumstances
1. To cure certain malignancies
2. To palliate symptoms in patients with disseminated cancer when the potential benefits
of treatment exceed the side effects of treatment
3. To treat asymptomatic patients
A. When the cancer is aggressive and treatable ( acute leukemia small cell lung
cancer)
B. When treatment has been proved to decrease the rate of relapse and increase the
disease –free interval or increase the absolute survival (stages 1 or 11 breast
carcinoma and estrogenic sarcoma)
4. To allow less mutilating surgery by treating first with chemotherapy alone or in
combination with radiotherapy
Classification of anti- cancer drugs
1.Alkylating agents
A. Nitrogen mustards
Mechlorethamine, cyclophosphamide,Chlorambucil, Melphalan
B. Ethyleminies
Thio-TEPA
C. Alkyl sulfonate
Busulfan
D. Nitrosoureas
Carmusstine, Lamustine, Samustine
E. Triazine
Dacarbazine
11. Anti-metabolites
A. Folate antagonist
Methotrexate
B. Purine antagonist
6- Marcapto-purine
C. Pyrimidine antagonist
5-Fluroro-uracil, cytarabine
111. Antibiotics
Doxorubicin , Daunorubicin, Dactinomycin , Bleomycin, Mitomycin-C,
Mitoxantrone
1V. Vinca alkaloids
Vincristine, Vinblastin
V .Taxanes
Paclitaxel, Docetaxel
V1. Epi-podophyllotoxin
Etoposide
V11 . Enzymes
L- Asparaginase
V111 . Radioactive isotopes
Radioiodine, Radio gold, Radio phosphorus
1X . Hormones
Glucocorticoids, Oestrogens, Antioestrogens, Progestins, Androgens, Antiandrogens,
X. Miscellaneous
Hydroxyurea, Cisplatin, Carboplatin, , procarbazine, Mitotane Tretinoin


1.Alkylating agents
E.g
Mechlorethamine, cyclophosphamide,Chlorambucil, Melphalan , Busulfan
Mechanism of action
Alkylating agents compounds contain chemical groups that can form covalent bonds with particular nucleophilic substances in the cell
In neutral or alkaline solutions , they undergo intermolecular cyclization to form a highly reactive ethylene imonium cation (quaternary ammonium compound )and releasing the chlorine
The strained ring of the ethylene imonium intermediate opens to form a reactive carbonium ion
Such ions are highly reactive and react instantaneously with an electro donor such as amine, hydroxyl or sulfhydroyl groups
Most of the cytotoxic anticancer alkylating agents are bifuctional means they have two alkylatng groups
The nitrogen at position 7 (N7) of guanine, being strongly nuclophilic, is probably the main molecular target for alkylaton in DNA to give 7-alkyguanine
The N7 being converted to a quaternary ammonium nitrogen
These reaction can then be repeated with the other CH2 CH2 Cl to give a crosslink
A bifuctuional agent ,being able to react with two groups, can cause intra or inter-chain cross-linking
Cross linking can cause defective replication through pairing of alkyl guanine and thymine, leading to substitution of AT for GC or it can cause excision of guanine and chain breakage
This can interfere not only with transcription but also with replication which is probably the critical effect of anticancer alkylating agents


Mechanism of alkylation of DNA guanine. A bis(chloroethyl)amine forms an ethyleneimonium ion and a carbonium ion that react with a base such as N7 of guanine in DNA, producing an alkylated purine. Alkylation of a second guanine residue, through the illustrated mechanism, results in cross-linking of DNA strands
Cyclophosphamide
Mechanism of action
This is a nucleophilic alkylating agent which inhibits the DNA synthesis
Metabolism
Native drug is inactive and requires activation by liver to form an aldehyde that decomposes in plasma and peripheral tissues to yield acrolein and an alkylating metabolite ( Phosphoramide mustard)
The liver also metabolizes to metabolites ti inactive compounds
Drugs that induce microsomal enzymes (ex – barbiturates ) may enhance toxicity , liver disease may decrease toxicity
Active and inactive metabolites are excreted in urine
Administration
The drug should be administered with a large volume of fluid in the morning or early afternoon to avoid cystitis
Dose modification may be required for hepatic or renal function impairment
Drug interactions with warfarin are known to prolong the prothrombin time and with succinyl choline to increase neuromuscular blockade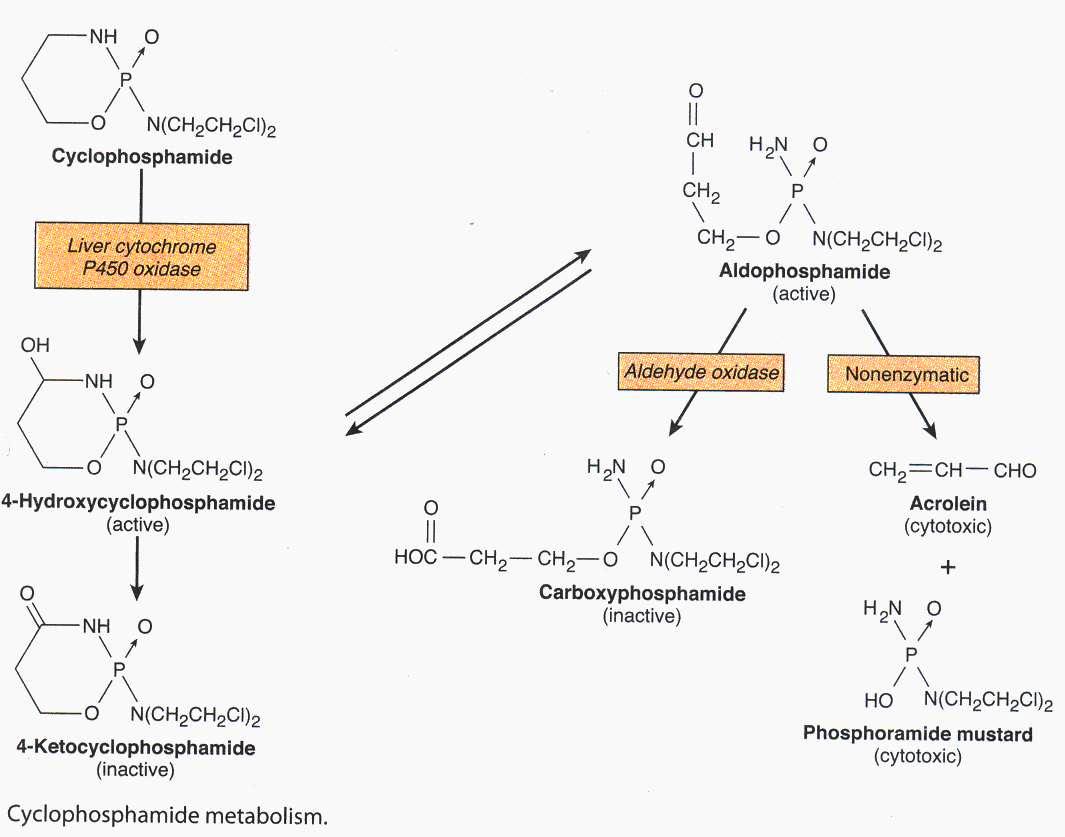
Toxicity
Alopecia
Bone marrow depression resulting in thrombocytopenia
Hepatotoxicity
Cystitis and bladder carcinoma
Preparation
Tab – 50 mg
Injections -100mg/gial, 200mg /vial, 599mg/vial
Dosage
Cyclophosphamide is frequently employed as part of combination chemotherapy regimens. Some common doses are 0.5 g/m2 intravenously every 3 weeks or 50- 200 mg/m2 orally for 4 days every 28 days
Uses
Autoimmune disorders like SLE, rheumatoid arthritis , Wagener’s granulomatosis and other systemic vasculitis
Ifosfamide
Mechanism of action
Mechanism is alkylation and DNA cross-linking and chain breakage.
Metabolites are similar to cyclophosphamide but not cross-resistant
This is a nucleophilic alkylating agent which inhibits the DNA synthesis
Metabolism
Inactive until activated by hepatic microbial enzymes like cyclophosphamide, the drug undergoes hepatic activation to an aldehyde form that decomposes in plasma and peripheral tissues to yield acrolein and its alkylation metabolites
Acrolein is highly toxic to urothelial mucosa
The chloroacetaldehyde metabolite may be responsible for much of the neurotoxic effects, particularly in patients with renal dysfunction.
Drug metabolites are excreted in urine
Toxicity
Alopecia
Nausea , vomiting, Neurotoxicity
Dizziness, confusion, ataxia & coma
Preparation
Injections -1g vial
Dosage
1000 – 1,200 mg/m2 intravenously over 30 minutes for 5 days every 3 – 4 weeks
Uses
Lymphomas, sarcomas and relapsed testicular carcinoma
Nitrosoureas
E. g
Carmusstine, Lamustine, Samustine

Mechanism of action
Mechanism is alkylation of DNA and RNA
DNA crossing ,inhibition of DNA polymerase, DNA repair and RNA synthesis
Metabolism
It is highly lipid= soluble drugs that enter the brain. Rapid spontaneous decomposition to active and inert products , the drugs are also metabolized. Most of the intact drugs and metabolic products are excreted in urine
Toxicity
Dose limiting
Myelosuppressions prolonged, cumulative and substantially aggravated by concurrent radiation therapy
Adverse effects
Nausea and vomiting can last up to 8 24 hours
These causes local pain during injection or it can also cause hypotension if injected too rapidly. It can also cause stomatitis , diarrhea, alopecia
Administration
It is supplied as 100 mg vials or 10, 40 and 100 mg capsules
Dosage
Carmustine - 150 – 200 mg/m2 intravenously every 6-8 weeks
Lomustine - 100 – 130 mg/m2 orally every 6 – 8 weeks
Uses
Brain cancer, lymphomas, multiple myeloma, melanoma and some carcinomas



11. Anti-metabolites
A. Folate antagonist
Eg- Methotrexate
Methotrexate
Methotrexate
Mechanism of action
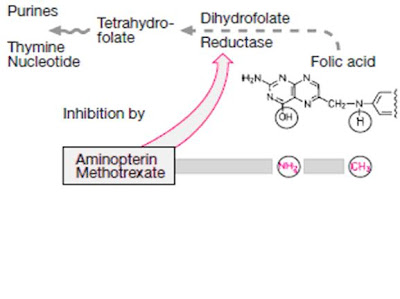
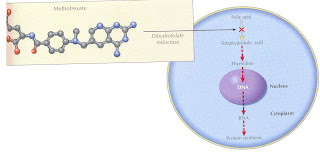
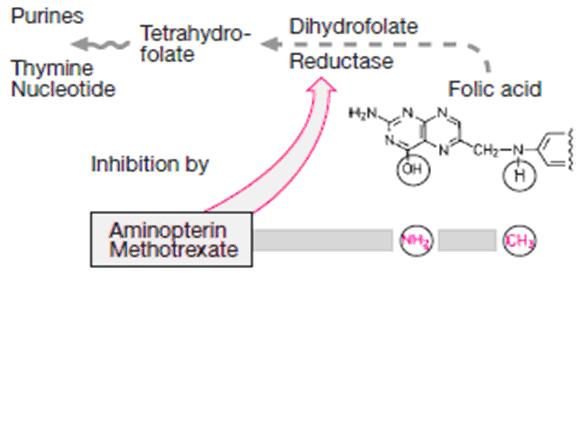
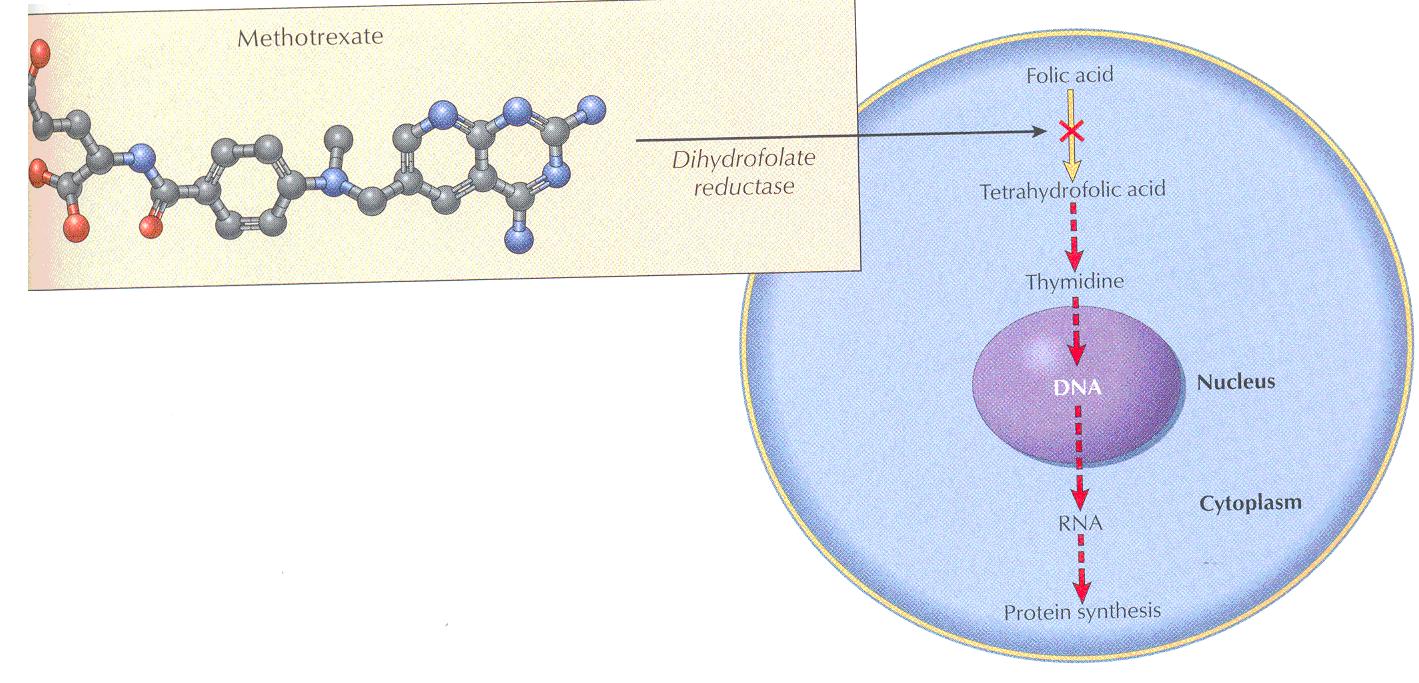
Methotrexate (MTX) is a folic acid antagonist that binds to the active catalytic site of dihydrofolate reductase (DHFR), interfering with the synthesis of the reduced form that accepts one-carbon units.
Folic acid is required in the synthesis for thymidylate (pyrimidine) and purine nucleotides and for DNA synthesis
Methotrexate resembles folic acid and competes with it at the active site of the enzyme dihydro folate recuctase (DHFR)
The affinity of methotrexate for this sites is 100000 times greater than that of dihydrofolate
The enzyme binds methotrexate with high affinity, and at pH 6.0, virtually no dissociation of the enzyme-inhibitor complex occurs .
At physiologic pH, reversible competitive kinetics occur
By blocking this step, methotrexate prevents nucleic acid synthesis and causes cell death
Metabolism
It is given orally, IV or intrathecally.
50 – 70% OF methotrexate is bound to plasma protein particularly albumin and alterations in plasma binding affect the pharmacokinetics of the drug
It is partly metabolized by the gut flora
About 80 – 95 % of the drug finally undergoes renal excretion either unchanged or as metabolites
Dose
Methotrexate Tab - 2.5 mg,
Inj – 5 ml
Drug interactions
Probeneid and salicylate increase methotrexate toxicity by competing for renal tubular secretion while simultaneously displacing it from plasma albumin binding sites
Furosemide and high dose vitamin C also block renal excretion
Gentamicin and cisplatin increase the toxicity of methotrexate by compromising renal excretion
Drug adverse effects
Nausea, vomiting , diarrhea , dermatitis , excessive lacrimation renal tubular necrosis, renal dysfunction liver cirrhosis &osteoporosis
Uses
It is used in the treatment of acute leukaemias, breast cancer and soft tissue sarcomas
It is also used in rheumatoid arthritis and psoriasis

B. Purine antagonist
6- Marcapto-purine, 6-Thioguanine, Allopurinol

Mercaptopurine (6-MP)

Mercaptopurine (6-MP)
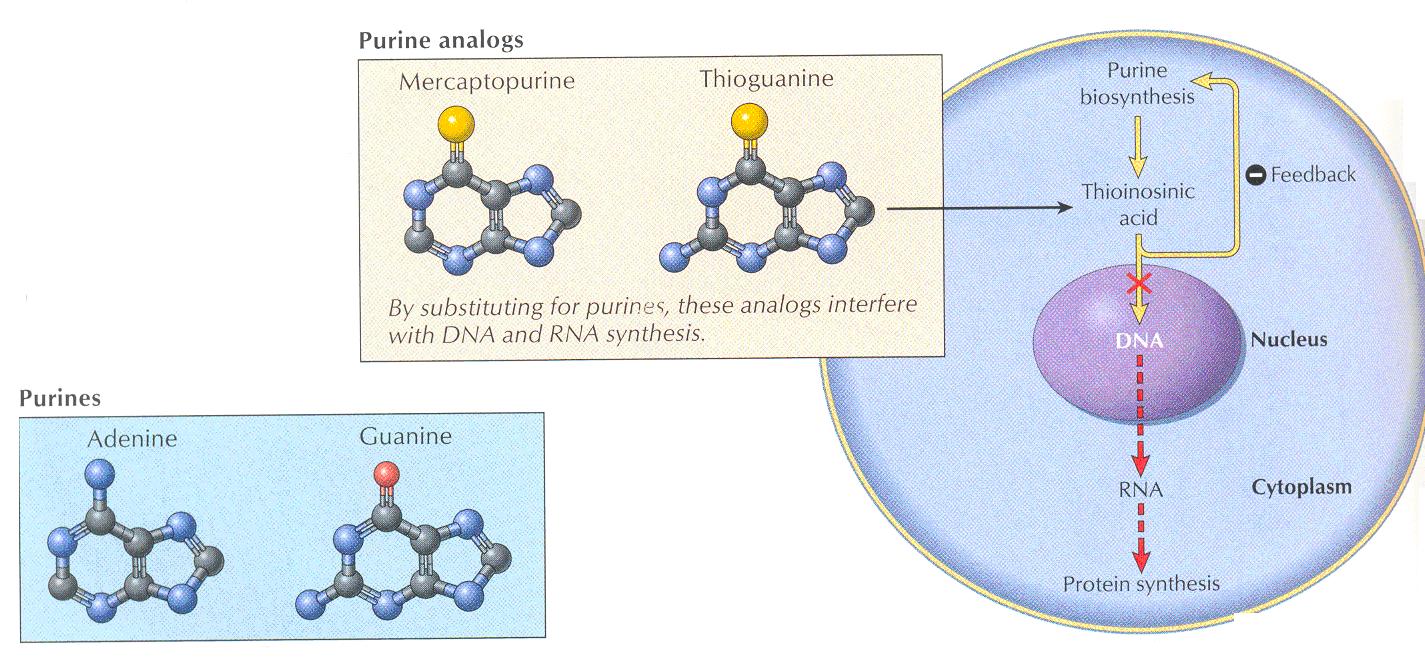
It was the first of the thiopurine series found useful as an anticancer drug.
Mechanism of action
6-MP, 6-TG and azathioprine are structural analogues of hypoxanthine and are converted by hytpoxanthine and are converted by hypoxanthine- guanine phosphoribosyl transfere into the ribonucleotides which inhibit biosynthesis of adenylic and guanylic acid from inosinic acid and thereby suppress DNA synthesis
Like other thiopurines, it must be metabolized by hypoxanthine-guanine phosphoribosyl transferase (HGPRT) to the nucleotide form (6-thioinosinic acid), which in turn inhibits a number of the enzymes of purine nucleotide interconversion.
Pharmacokinetics
They are well absorbed from the gut, metabolized by xanthine oxides in the liver and excreted as metabolites
Allopurinol inhibits xanthine oxidase and increases the toxicity
Mercaptopurine is used primarily in the treatment of childhood acute leukemia, and a closely related analog, azathioprine, is used as an immunosuppressive agent
Drug interaction
Anticoagulant action of warfarin may be inhibited by mercaptopurine. Effect influenced by allopurinol
Dose
2.5 mg/kg/day orally
Adverse drug reaction
Bone marrow depression is the chief toxicity . Side effects also include nausea, vomiting , diarrhea& hepato toxicity
Therapeutic uses
6- MP is given orally for the treatment of acute lymphatic leukemia and in chronic myeloid leukemia refractory to busulfan
Thioguanine (6-TG)
Mechanism of action
It inhibits several enzymes in the purine nucleotide pathway.
A variety of metabolic lesions are associated with the cytotoxic action of the purinethiols.
These include inhibition of purine nucleotide interconversion; decrease in intracellular levels of guanine nucleotides, which leads to inhibition of glycoprotein synthesis; interference with the formation of DNA and RNA; and incorporation of thiopurine nucleotides into both DNA and RNA.
Dose
2 mg/kg/day orally
Adverse effects
Myelo- suppression, immunosuppressant & hepatotoxicity
Uses
6-TG has a synergistic action when used together with cytarabine in the treatment of adult acute leukemia.
Azathioprine
Suppresses cell mediated immunity and is used mainly for immunosuppression in rheumatoid arthritis , organ transplantation
Adverse effects
In adddition to those decreined earlier , hyper- uricaemia may occor
C. Pyrimidine antagonist
E.g
5-Fluroro-uracil, cytarabine
5- Fluroro –uracil is a fluorine substituted analogue of uracil
Cytarabine is an analogue of cytidine in which robose has been replaced by arabinose

5- Fluorouracil
5-Fluorouracil (5-FU) is a prodrug and undergoes a complex series of biotransformation reactions to ribosyl and deoxy - ribosyl nucleotide metabolites.

Pharmacokinetics
Fluorouracil is normally given intravenously and has a short metabolic half-life on the order of 15 minutes.
It is not administered by the oral route because its bioavailability is erratic due to the high levels of the breakdown enzyme dihydropyrimidine dehydrogenase present in the gut mucosa.
Dose
15mg/kg/d IV for 5 days by 24 hrs infusion. 15 mg/kg weekly 1V
Drug interactions
May increase the waffarin action. Its action may be reduced by allopurinol
Adverse effects
Nausea, vomiting, diarrhea, alopecia, bone marrow depression, & anorexia
Uses
A cream incorporating fluorouracil is used topically for treating basal cell cancers of the skin.
Fluorouracil is the most widely used agent for the treatment of colorectal cancer, both as adjuvant therapy as well as for advanced disease.
In addition, it has activity against a wide variety of solid tumors, including cancers of the breast, stomach, pancreas, esophagus, liver, head and neck, and anus
Cytarabine
Mechanism of action
Cytarabine is an S phase-specific antimetabolite that is converted by deoxycytidine kinase to the 5'-mononucleotide (AraCMP).
AraCMP is further metabolized to the triphosphate (AraCTP), which competitively inhibits DNA polymerase ,blocks the generation of cytidilic acid and results in blockade of DNA synthesis.
Cytarabine is also incorporated into RNA and DNA.
Pharmacokinetics
After intravenous administration, the drug is cleared rapidly, with most being deaminated to an inactive form.
The ratio of the anabolic enzyme deoxycytidine kinase to the inactivating catalyst cytidine deaminase is important in determining the cytotoxicity of cytarabine.
In view of cytarabine's S phase specificity, the drug is highly schedule-dependent and must be given either by continuous infusion or every 8–12 hours for 5–7 days.
Its activity is limited almost entirely to treatment of acute myelogenous leukemia, for which it is a major drug
Dose
100 mg/m2 by continuous infusion given for 5 – 10 days
Drug interaction
Reduces plasma concentration of digoxin , antagonizes Gentamycin
Drug adverse effects
Nausea , vomiting, alopecia , bone marrow depression ,stomatitis & cerebral ataxia
Uses
It is used in the treatment of acute leukemia , Refractory disease & leukaemic meningitis
Gemcitabine
Gemcitabine is phosphorylated initially by the enzyme deoxycytidine kinase and then by other nucleoside kinases to the di- and triphosphate nucleotide forms, which then inhibit DNA synthesis. 
Mechanism of action
It is a synthetic pyrimidine nucleoside and cytarbine analogue
It is metabolized intravellularly to active diphosphate and triphosphate nucleosides
It inhibits DNA synthesis and induce apoptosis and is primarily active against cells in the S- phase but may also cause cellular arrest at the G1 – S border
Pharmacokinetics
It is infused IV . It is deaminated to difluoro-deoy-uridie, which is not cytotoxic
It is excreted in urine
Dose
1000mg /m2 IV by 30 mints infusion on days 1,8 and 15 of each 28 day cycle
Drug interactions
With Alcohol produces GI irritation
Adverse effects
Myelo- suppression is the dose – limiting toxicity of gemcitabine
Other toxicities include nausea, vomiting , alopecia, rash and flu like syndrome
Uses
Gemcitabine was initially approved for use in pancreatic cancer but is now widely used in the treatment of non-small cell lung cancer and bladder cancer.
111. Antibiotics
The anthracycline antibiotics, isolated from Streptomyces peucetius var caesius, are among the most widely used cytotoxic anticancer drugs.

Several other anthracycline analogs have entered clinical practice, including idarubicin, epirubicin, and mitoxantrone.
Daunorubicin was the first agent in this class to be isolated, and it is still used in the treatment of acute myeloid leukemia.
Doxorubicin has a broad spectrum of clinical activity against hematologic malignancies as well as a wide range of solid tumors.
The entire class of anthracyclines exert their cytotoxic action through four major mechanisms.
These are
(1). Inhibition of topo - isomerase II;
(2). High-affinity binding to DNA through intercalation, with consequent blockade of the
synthesis of DNA and RNA
(3). Binding to cellular membranes to alter fluidity and ion transport;
(4) Generation of semiquinone free radicals and oxygen free radicals through an enzyme-
mediated reductive process.


Doxorubicin
It is a cytotoxic anthracycline antibiotic
Doxorubicin is the hydroxylated analog of daunorunicin
Doxorubicin is one of the most important and widely used anticancer drugs
Doxorubicin has a broad spectrum of clinical activity against hematological malignancies as well as a wide range of solid tumors.
Mechanism of action
The cytotoxic action results from it s binding to DNA and inhibition of nucleic acid synthesis

Pharmacokinetics
In the clinical setting, anthracyclines are administered via the intravenous route .
The anthracyclines are metabolized extensively in the liver, with reduction and hydrolysis of the ring substituents
The hydroxylated metabolite is an active species, whereas the aglycone is inactive.
Up to 50% of drug is eliminated in the feces via biliary excretion, and for this reason dose reduction is required in the setting of liver dysfunction.
Dose
Treatment of metastatic breast carcinoma
Adult – 60 – 75 mg/m2 once every 3 weeks in combination with cyclophosphamide given as an infusion over 1 hr diluted in 0.9% sodium chloride or 5% glucose
Reduce dose to 50% in patients with moderate liver dysfunction
Drug interactions
Doxorubicin interacts with a number of other drugs
E.g. – Amino glycosides, steroids, aminophyline and propranolol
Drug adverse effects
Leucopoenia, thrombocytopenia, nausea , vomiting , diarrhea
Uses
Doxorubicin is one of the most important anticancer drugs, with major clinical activity in
carcinomas of the breast, endometrium, ovary, testicle, thyroid, stomach, bladder, liver, and lung;
Daunorubicin
It is a cytotoxic anthracycline antibiotic
Daunorubicin has a far narrower spectrum of activity than doxorubicin
Mechanism of action
It forms a stable complex wit DNA and interferes with the nucleic acid synthesis
It is a cell – cycle nonspecific effects are mostly marked to cells in the s- shape
It also has immunosuppressant and antibacterial effects
Pharmacokinetics
In the clinical setting, anthracyclines are administered via the intravenous route .
The anthracyclines are metabolized extensively in the liver, with reduction and hydrolysis of the ring substituents
The hydroxylated metabolite is an active species, whereas the aglycone is inactive.
Up to 50% of drug is eliminated in the feces via biliary excretion, and for this reason dose reduction is required in the setting of liver dysfunction.
Dose
Adult – 30 – 45 mg/m2 body surface daily for 2-3 days as a solution in 0.9% sodium chloride administered into a fast running infusion for sodium chloride or glucose
Repeated after 3 – 6 weeks
Child 25mg/m2 once a week in combination with other regiments
Drug interactions
Incompatible with heparin , aluminum and dexamethasone
Immunization with live vaccines is not recommended as daunorubicin is an immunosuppressant
Drug adverse effects
Nausea, vomiting, diarrhea, stomatitis, alopecia and dermatological reactions
Use
Daunorubicin has been mainly used for the treatment of acute lymphoblastic leukemia,
Dactinomycin
Dactinomycin is an antitumor antibiotic isolated from a Streptomyces organism.
Mechanism of action
It binds tightly to guanine portion of DNA forming a complex which interferes with DNA and RNA synthesis as well as protein synthesis
Dose
Dactinomycin is mainly used to treat pediatric tumors such as Wilms' tumor, and Ewing's sarcoma
Adult - 2.5 mg/m2 in divided doses over 1 week, repeated every 2 weeks or 400-600 mcg/m2 /daily for 5 days, repeated every 3- 6 weeks
Child more than 6 months 15 mcg/kg/day or 400 – 600 mcg /m2/day for 5 days every
3 – 6 weeks
Drug interactions
It may interfere with bioassay bioassay procedures for the determination of antibacterial drug levels
Drug adverse effects
Nausea, vomiting, diarrhea, stomatitis, GIT ulceration, anorexia, hepatitis, anemia , alopecia, leucopoenia & thrombocytopenia.
Uses
Dactinomycin is mainly used to treat pediatric tumors such as Wilms' tumor, and Ewing's sarcoma
Mitomycin

Mitomycin is an antibiotic isolated from Streptomyces caespitosus.
Mechanism of action
It suppresses nucleic acid synthesis
It is a non- specific cell- cycle agent but has marked action in G1 and early S phases
Mitomycin when converted to an active metabolite , inhibits DNA synthesis as also that of RNA and protein
May be selectively toxic to hypoxic cells because it may generate super oxide and hydroxyl radical
Effects are most marked in the late G1 and early S phases of cell cycle
Dose
Adult - Initially, 10 – 20 mg/m2 body surface area , repeated at intervals of 6 – 8 weeks
Drug interactions
Synergism with 5- fluorouracil. Enhances cytotoxic effect of radiation especially in the presence of hypoxia
Drug adverse effects
Nausea, vomiting, diarrhea, stomatitis, GIT ulceration, anorexia, hepatitis, anemia , alopecia, leucopoenia & thrombocytopenia.
Uses
It is used in the treatment of solid tumors including breast, cervix, eye, liver, lung and stomach
One special application of mitomycin has been in the treatment of superficial
bladder cancer.
.
Mitoxantrone
Mitoxantrone is an anthracene compound whose structure resembles the anthracycline ring.
Mechanism of action
It binds to DNA and inhibits topoisomerase it causing DNA strand breakage leading t decreased cell replication..
Pharmacokinetics
The plasma half-life of mitoxantrone in patients is approximately 75 hours, and it is predominantly excreted via the hepatobiliary route in feces
Dose
Treatment of acute leukemia - 8 – 12 mg/m2/day, once daily for 4 – 5 days
Treatment of breast prostate cancer and lymphomas - Adult – 12 -14 mg/m2/day for 5 days every 4 weeks .
Drug interactions
It appears to have synergistic activity with 5 – fluorouracil , vincristine,dacarbazine and methotrexate in animal studies
Drug adverse effects
Nausea ,vomiting, diarrhea, alopecia, stomatitis, fever , GI bleeding ,fatigue and blue – green urine discoloration.
Uses
Bleomycin

Mechanism of action
Bleomycin is a small peptide that contains a DNA-binding region and an iron-binding domain at opposite ends of the molecule.
It acts by binding to DNA, which results in single-strand and double strand breaks following free radical formation, and inhibition of DNA biosynthesis.
The fragmentation of DNA is due to oxidation of a DNA-bleomycin-Fe(II) complex and leads to chromosomal aberrations.
Bleomycin is a CCS drug that causes accumulation of cells in the G2 phase of the cell cycle.
Pharmacokinetics
One advantage of this agent is that it can be given subcutaneously, intramuscularly, or intravenously
Peak blood levels of bleomycin after intramuscular injection appear within 30–60 minutes.
Intravenous injection of similar dosages yields higher peak concentrations and a terminal half-life of about 2.5 hours
Elimination of bleomycin is mainly via renal excretion; for this reason, dose modification is recommended in the setting of renal dysfunction.
Dose
Treatment of squamous cell or testicular tumors
Adult – 15000 IU – 3 times a week or 30,000 twice a week IM/IV , repeated at usual intervals of 3 – 4 weeks up to a total cumulative dose of 500,000 IU
Treatment of lymphoma
Adult – 15000 IU – 3 times a week or 30,000 twice a week IM/IV , repeated at usual intervals of 3 – 4 weeks up to a total cumulative dose of 225,000 IU
Drug interactions
Lomustine increases severity of leucopenia
Elimination decreased by cisplatin
Decreases plasma levels of digoxin and phenytoin
Drug adverse effects
Fever , thrombophloebitis, hyperpyrexia, stomatitis, alopecia, and hyper pigmentation.
Uses
Bleomycin is indicated for the treatment of Hodgkin's and non-Hodgkin's lymphomas, germ cell tumor, head and neck cancer, and squamous cell cancer of the skin, cervix
1V. Vinca alkaloids
Ex- Vincristine & vinblastine
Mitotic spindles
The mitotic inhibitors act by interfering with the mitosis of cells and thereby inducing mitotic arrest
The mitosis takes lace during the M phase of the cell cycle
Mitotic arrest is induced because of damage to the spindle apparatus
The chromatides , which are separated in the metaphase are prevented from being pulled toward opposite poles in the following anaphase
The separate threads of the spindle apparatus are built as a microtubule system
Microtubules are hollow , cylindrical structures built up of the protein tubulin , which consists of two subunits, alpha and beta – tubulin with very similar amino acid sequences
The cylinder consists of 13 rows of tubulin heterodiamers, the protofilaments
The microtubule cylinder can also be regarded as consisting of a helical array of altering arrays of tubulin subunit
Some of the drugs have a common binding site and others a different, but probably common, binding site and others a different but probably common , binding site
Taxanes promote the assembly of tubulin to microtubules and prevent depolymerization by biding to tubulin at separate site
Microtubules are part of the cytoskeleton and take part in intracellular transport and communication
Some of the toxic side effects of the mitotic inhibitors may be due to disturbance of these phenomena


VInblastine
Vinblastine is an alkaloid derived from Vinca rosea, the periwinkle plant.
Mechanism of action
Its mechanism of action involves depolymerization of microtubules, which are an important part of the cytoskeleton and the mitotic spindle.
Binds to micro tubular proteins . It inhibits RNA synthesis by affecting DNA-dependent RNA polymerases.
Cell cycle- phase specific , it arrests cells at the G2 phase and M phase interface
Pharmacokinetic
Highly bound to plasma proteins.
It is metabolized by the liver to active and inactive metabolites
It is predominantly excreted in bile
Minimum free drug is recovered in urine
Drug interactions
Mitomycin C, phenytoin , alpha interferon
Adverse effects
Toxicity includes nausea , vomiting, diarrhea, bone marrow suppression, and alopecia.
Dosage
30mg/m2 IV every weekly
Uses
It has clinical activity in the treatment of Hodgkin's disease, non-Hodgkin's lymphomas, breast cancer, and germ cell cancer.
VIncristine
Vincristine is also an alkaloid derivative of Vinca rosea and is closely related in structure to vinblastine.
Mechanism of action
Vincristine arrests cell division at the metaphase stage by inhibiting microtubule formation in the mitotic spindle
Drug interactions
It may decrease digoxin and phenytoin serum levels .
Synergistic with methotrexate.
Adverse effects
The main dose limiting toxicity is neurotoxicity.
While myelosuppression can occur, it is generally milder and much less significant than with vinblastine.
Dose
1.5 mg/m2 IV . Maximum 2 mg weekly
Uses
Vincristine has been effectively combined with prednisone for remission induction in acute lymphoblastic leukemia in children.
V . Taxanes
Eg- Paclitaxel and docetaxel
The mitotic spindle is part of larger , intracellular skeleton (cytoskeleton) that is essential for the movements of structures occurring in the cytoplasm of all eukaryotic cells
The mitotic spindle consists of - chromatin + a system of microtubules compose of the protein tubulin
The mitotic spindle is essential for the equal partitioning of DNA into the two daughter cells that are formed when a eukaryotic cell divides .
Several plant –derived substances used as anticancer drugs disrupt this process by affecting the equilibrium between the polymerized and depolymerized forms of the microtubules , thereby causing cytotoxicity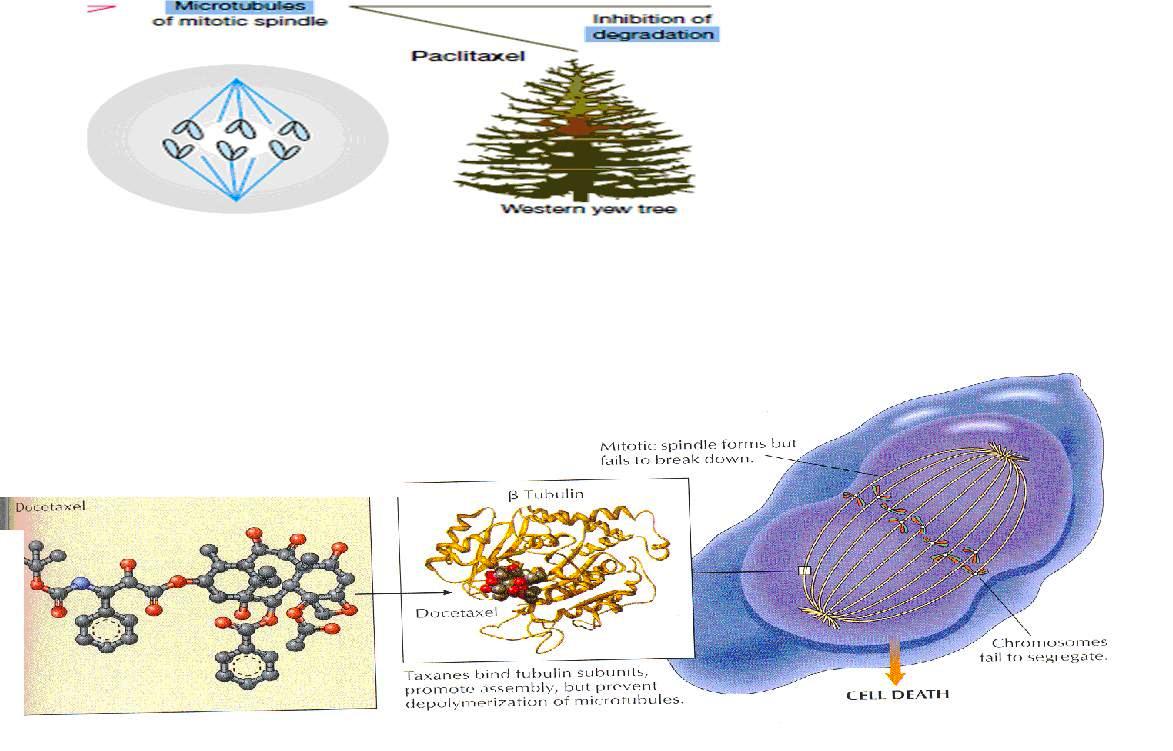
Paclitaxel
Better known as taxol, paclitaxel is the first member o the taxane family to be used in cancer chemotherapy
It is an alkaloid ester derived from the Western yew (Taxus brevifolia) and the European yew (Taxus baccata).

Mechanism of action
(Taxanes bind tubulin subunits promote assembly but prevent depolymerization of microtuubles )
These are active in the G2 / M phase for the cell cycle
They bind reversibly to the BETA – tubulin subunit , but unlike the vinca alkaloids, they promote polymerization and stabilization of the polymer rather than disassembly
They shift the depolymrizaiton- polymerization process to favor the formation of microtubules
This promotion of microtubule assembly by paclitaxel occurs in the absence of microtubule-associated proteins and guanosine triphosphate and results in inhibition of mitosis and cell division.
The overly stable microtubules formed are nonfunctional and chromosome desegregation does not occur . This results in death of the cell
Pharmacokinetics
It have a large volume of distribution but not enters the brain
It is metabolized extensively by the liver P450 system, and nearly 80% of the drug is excreted in feces.
Biliary excretion are responsible for their elimination into the stool
Dose modification is not required in patients with renal impairment but doses should be reduced in patients with hepatic dysfunction
Adverse effects
The dose – limiting toxicity of paclitaxel is neutropenia
Hypersensitivity reactions may be observed in up to 5% of patients, but the incidence can be reduced by premeditation with dexamethasone, diphenhydramine, and an H2 blocker.
Uses
Paclitaxel has significant activity in a wide variety of solid tumors, including ovarian, advanced breast, non-small cell and small cell lung, head and neck, esophageal, prostate, and bladder cancer.
Docetaxel
It is a semi synthetic taxane derived from the European yew tree.
Its mechanism of action, metabolism, and elimination are identical to those of paclitaxel. It is approved for use as second-line therapy in advanced breast cancer and lung cancer, and it also has major activity in head and neck cancer, gastric cancer, ovarian cancer, and bladder cancer. V1. Epi -podophyllotoxins
Two compounds, VP-16 (etoposide) and a related drug, VM-26 (teniposide), are semi synthetic derivatives of podophyllotoxin, which is extracted from the may apple root (Podophyllum peltatum).
Both an intravenous and an oral formulation of etoposide are approved for clinical use in the USA.
Mechanism of action
Etoposide and teniposide are similar in chemical structure and in their effects
They block cell division in the late S-G2 phase of the cell cycle.

Phgarmacoketics
These agents are administered via the intravenous route
These are rapidly and widely distributed throughout the body except for the brain.
Up to 90–95% of drug is protein-bound, mainly to albumin.
Dose reduction is required in the setting of renal dysfunction
Dosage
Etoposide is given in combination with other antineoplastic drugs
50 -100 mg /m2/day from days 1 -5 or 100 mg/m2 on days1,3 and 5
Adverse drug effects
Nausea. Vomiting , anorexia, diarrhea, stomatitis, alopoecis,,CNS defects
.
Uses
Etoposide has clinical activity in germ cell cancer, small cell and non-small cell lung cancer, Hodgkin's and non-Hodgkin's lymphomas, and gastric cancer and as high-dose therapy in the transplant setting for breast cancer and lymphomas.
V11 . Enzymes
E.g
Asparaginase
Asparaginase is an enzyme that is isolated from various bacteria mainly E. Coli for clinical use.
Mechanism of action
It hydrolyzes circulating L-asparagine to aspartic acid and ammonia.
Aspargine is a non- essential amino acid normally synthesized by the mammalian tissue cells
Depletion of L-asparagine results in effective inhibition of protein synthesis
Certain malignant tumors are unable to synthesize aspargine and are dependent on supplies from the host
It acts by depleting asparagines from the host , hydrolyzing circulating l- aspargine to aspartic acid denying the malignant cells the essential metabolite
It is relatively nontoxic to normal cells
Administration
The enzyme must be administered either IV or intramuscularly because it is destroyed by gastric enzymes
It may cause hypersensitivity pyrogenic reactions and skin rashes
Adverse effects
The main adverse effect of this agent is a hypersensitivity reaction manifested by fever, chills, nausea and vomiting, skin rash, and urticaria.
Severe cases can present with bronchospasm, respiratory failure, and hypotension.
and neurological toxicity with confusion, hallucinations, and coma.
Dose
20,000 IU /m2 daily IV for 5 – 10 days

VIII . Radioactive isotopes
Radioiodine, Radio gold & Radio phosphorus
These are the isotopes of various elements which act because of their physical property of emitting radiation
Radiations given out produce ionization in the cells
The vital enzymes of cells are in activated and there occurs structural changes in the chromosomes
Ultimately there is cell - destruction
Radium
It is a bivalent meal . It disintegrates with emission of various types of radiations
It has been referred to as radioactive
There are three types of rays emitted by radium
Alpha rays
These consist of two protons and two neutrons . It has very little power of penetration tissues
Beta rays
These consist of negatively or positively charged , electros
They have more penetrating power than alpha - rays
Gamma rays
These are gamma electromagnetic waves of shortwave length and have greater power to penetrate the tissues
These waves cannot pass through elements of high atomic number
Eg – lead

Geigy – Muller counter
Radioactive isotopes are very sensitive and can be detected by Geigy – Muller counter, electroscopes an electro radiography
Clinically useful radioactive isotopes are Radioiodine , Raiophosphorous and Radio gold
1. Radioactive iodine (I131)
It is obtained as a fission product of uranium
It can also be prepared by neutron irradiation of tellurium. Sodium radio iodine (I131)
preparations are used orally as well as intravenously.
It acts by giving beta radiation
It is rapidly absorbed and taken up by the thyroid gland
There is no radiation sickness and does not necessitate repeated blood examination
Dose
It is given in a dose of 4- 10milli curies for thyrotoxicosis and 50-100 mill curies for thyrotoxicosis for thyroid carcinoma
Uses
It is used in hyperthyroidism ,& carcinoma of the thyroid gland for determining functional activity for the thyroid gland
2. Radio- active isotopes ( P 32)
It is used in the form of disodium hydrogen phosphate .
It is distributed to all parts of the body .
Uses
It is used in diseases like polycythemia vera and chronic lymphatic leukemia
Dose
It is given in a dose of 3 – 5 mill curies I.V
3. Radio active Gold (Au 198 )
It is a short lived isotopes( half life 2 – 7 days ).
It emits both beta and gamma rays .
Dose
35 – 100 mill curies is injected intrapleurally or intraperitoneally
Use
It is used in malignant pleural and peritoned effusions
1X . Hormones & their antagonists
Prednisone
It is a potent , synthetic , anti- inflammatory corticosteroid with less mineral corticoid activity than cortisol
The use of this compound in the treatment of lymphomas arose when it was observed that patients with cushing syndrome , which is associated with hyper secretion of cortisol, have lymphocytopenia and decreased lymphoid mass
Prednisone is primarily employed to induce remission in patients with acute lymphocytic leukemia and in the treatment of both Hodgkin and non- Hodgkin lymphomas
Mechanism of action
Prednisone itself is inactive and must first be reduced to prednisolone by 11- beta- hydroxysteroid dehydrogenase
This steroid then binds to a receptor that triggers the production of specific proteins
Pharmacokinetics
It is readily absorbed orally
Like other gluco-corticoids it is bound to plasma albumin and transcortin
It undergoes 11 – beta – hydroxylation to prednisolone in the liver
It is the active drug
The latter is glucuronidated and excreted into the urine along with the parent compound
Adverse effects
It has many of the adverse effects associated with glucocorticiids.
It can predispose to infection and to ulcers and pancreatitis
Other effects include hyperglycemia, cataract formation , glaucoma , osteoporosis, and change in mood
Dose
Prednisolone is generally started in doses of 60 – 100 mg daily in divided doses and them depending upon the response ,reduced to a maintenance dose of 20 – 40 mg daily
Estrogens
Estrogens, such as ethinyl estradiol or diethyl-stilbestrol had been used in the treatment of breast cancer
They have been largely replaced by the GnRH analogs because of fewer adverse effects
Estrogens inhibit the growth of prostatic tissue by blocking the production of LH, thereby decreasing the synthesis of androgens in the testis
Toxicity
Estrogen treatment can cause serious complications , such as hypertension t, MI , strokes thromboembolic, jaundice, retinal thrombosis disorder and hyjpercalcemia
Administration
It is supplied as 0.25, o.5, 1.0 & 5.0 mg tablets
Dosage
1 – 15 mg daily in divided doses
Use
Breast carcinoma
Estrogen antagonist
Eg - Tamoxifen, Toremifene, Fulvestrant
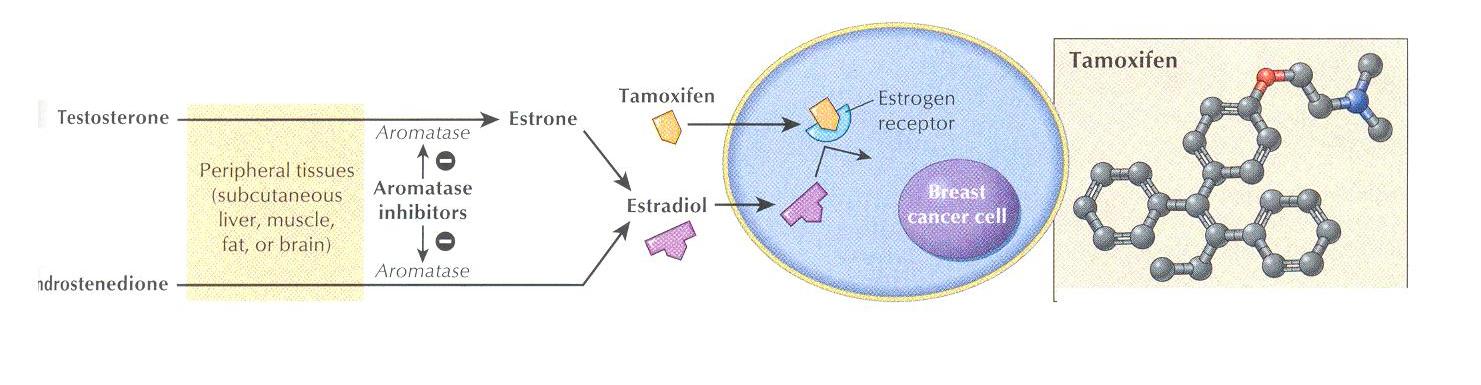

Tamoxifen is an estrogen antagonist
It is structurally related to the synthetic estrogen di- ethyl- stilbestrol and is used for first – line therapy in the treatment of estrogen receptor – positive breast cancer
It has weak estrogenic activity and is classified as a selective estrogen – receptor modulator
Mechanism of action
Tamoxifen & toremifene are nonsterioidal agents that bind to oestrogen receptors and may exert antioestrogenic, estrogenic or both activities
Fulvestrant is an oestrogen receptor antagonist without known agonist effects
Tamoxifen binds to the estrogen receptor, but the complex is not productive
The complex fails to induce estrogen – responsive genes and RNA synthesis does not ensue
The result is a depletion of estrogen receptors and the growth – promoting effects of the natural hormone and other growth factors are suppressed
Pharmacokinetics
Tamoxifen is effective on oral administration
It is partially metabolized by the liver
Some metabolites possess antagonist activity , whereas others have agonist activity
Unchanged drug and its metabolites are excreted predominantly through the bile into the feces
Adverse effects
It causes menstrual changes, vaginal discharge, uterine bleeding , thrombocytopenia, nausea , vomiting , skin rash , , hypocalcaemia & hair loss
Preparation
Tamoxifen - 10 mg, 20 mg tabs
Dosage
Tamoxifen - 20 mg orally once daily
Toremifene - 60 mg orally once daily
Fulvestrant - 250mg intramuscularly as a 5 ml solution monthly
Use
In treatment of breast carcinoma
Androgens (Fluoxymesterone and Methyl testosterone)
Therapeutic uses
Breast carcinoma, short- range anabolic effect and stimulation of erythropoiesis
Toxicity
It varies among preparations
Fluid retention , hepatotoxicity & hypocalcaemia
Administration
It issue with caution in patients with cardiac, hepatic or renal disease
Fluoxymesterone is administered in dose of 10 – 40 mg /day in 2- 4 divided doses and methyl-testosterone in dose of 50-200 mg/day in 2 or 3 divided doses
Anti- androgens
Ex - Flutamiide, Bicalutamide & Nilutamide
These are synthetic , non-steroidal antiandrogens used in the treatment of prostate cancer
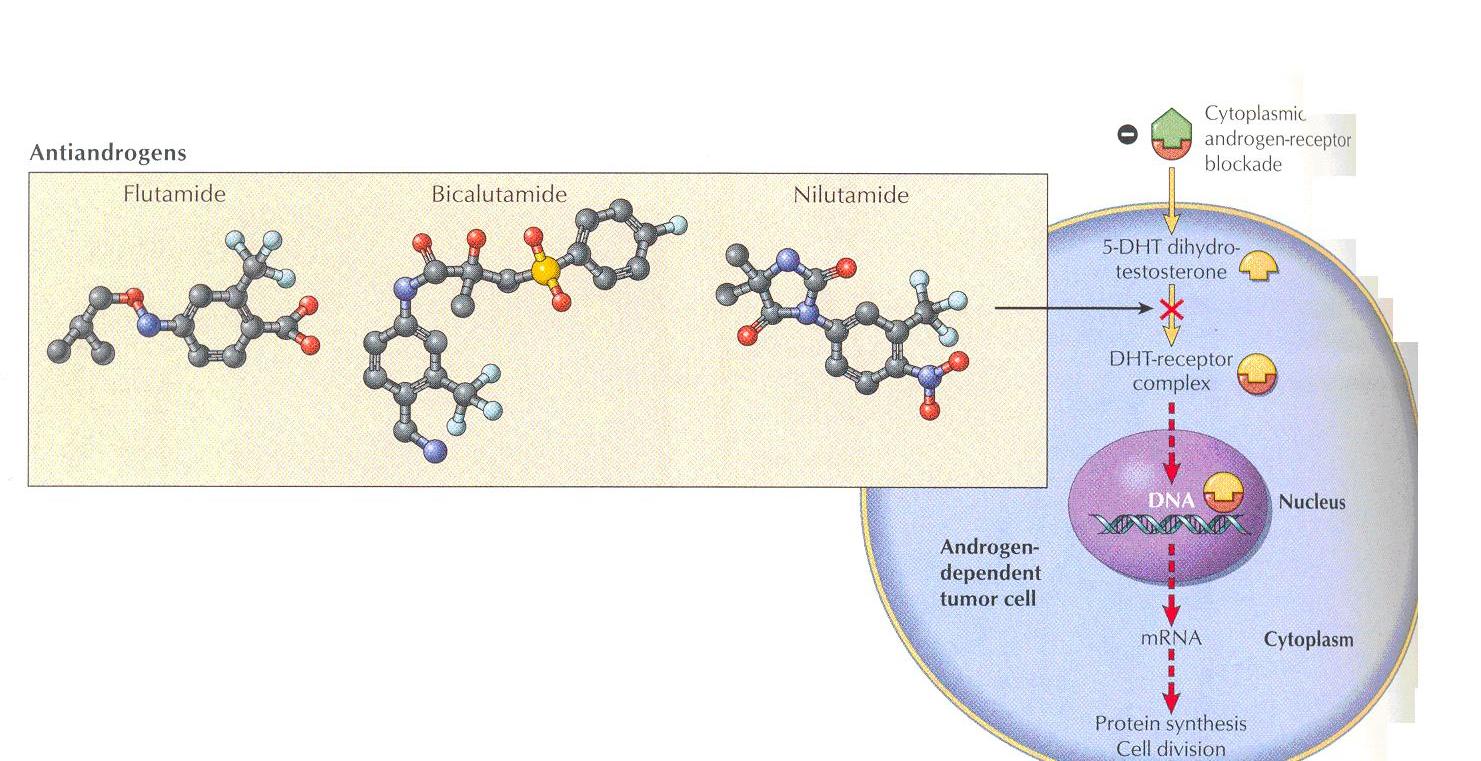
Mechanism of action
Nonsteroidal anti-androgens bind to cytosol androgen receptors and competitively inhibit the uptake or binding of androgens in target tissues
They compete with the natural hormone for binding to the androgen receptor and prevent its translocation into the nucleus
Flutamide is metabolized to an active hydroxyl derivative that bins to the androgen receptor
Flutamide blocks the inhibitory effects of testosterone on gonadotropin secretion causing and increase in serum LH and testosterone levels
Pharmacokinetics
Flutamide is always administered in combination with leuprolide or goserelin
These antiandrogens are taken orally
The drugs are almost totally metabolized
These agents are cleared through the kidney
Toxicity
Side effects include gynecomastia, GIT disturbances, impotence , diarrhea & liver failure in the case of flutamide .
It also causes nausea , vomiting , myalgia depression & mild hypertension
Nilutamide can cause visual problems
Administration
It is used with caution in patients with hepatic dysfunction
Flutamide - 250 mg divided dose orally
Bicalutamide - 50 mg orally once daily
Nilutamide - 300 mg once daily orally for 30 days, then 150 mg daily
Therapeutic uses
Prostate cancer , in combination with medical therapy
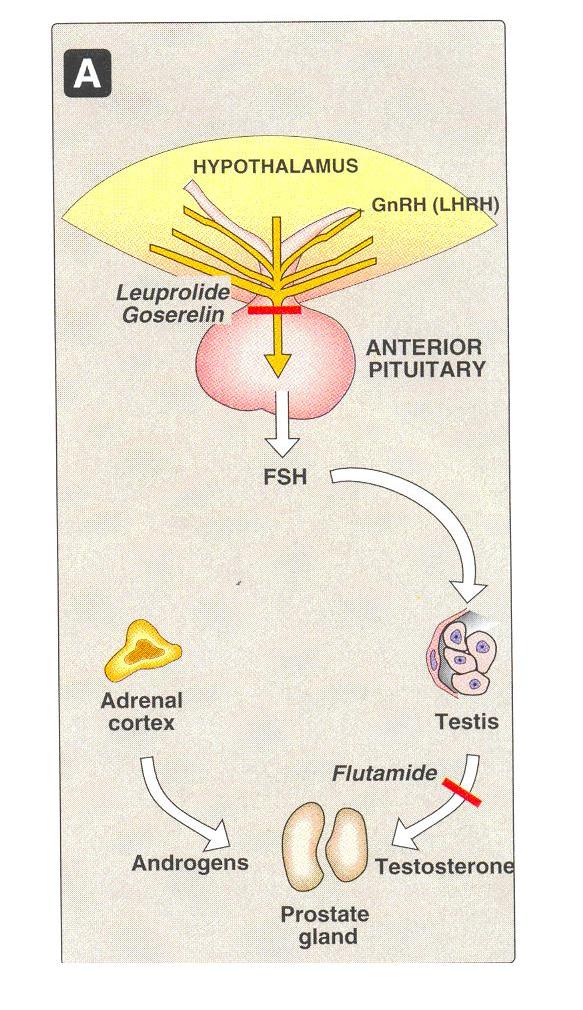
Gonadotropin – releasing hormone
Eg
Leuprolide & goserelin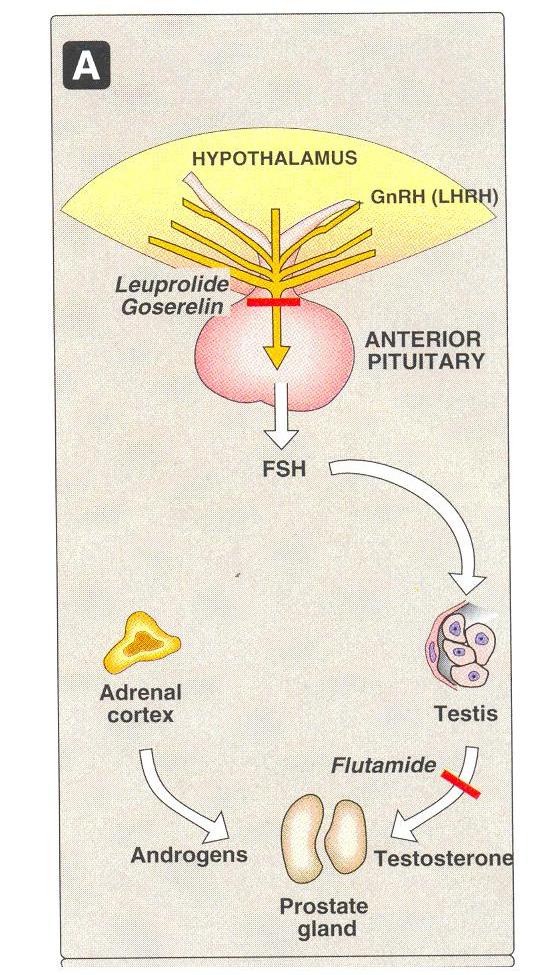
Leuprolide and goserelin
These are synthetic peptide analogs of naturally occurring gonadotropin releasing hormone (GnRH, LHRH).
Gonadotropin – releasing hormone (GnRH) is normally secreted by the hypothalamus
These stimulates the anterior pituitary to secrete the gonadotropic hormones, LH & FSH
The synthetic Nona peptides, leuprolide and goserelin are analogs of GnRH
Mechanism of action
As GnRH agonists they occupy the GnRH receptor in the pituitary , which leads to its desensitization and consequently , inhibition of release of FSH and LH
The both androgen and estrogen synthesis are reduced
Pharmacokinetics
Goserelin acetate is implanted intramuscularly
Levels of androgen may initially rise but then fall to castration levels
Adverse effects
The adverse effects of these drugs , including impotence, hot flushes & gynecomastia
Administration
Leuprolide and goserelin are now formulated in long-acting depot forms, which allows for administration once every 3 months.
Uses
Leuprolide and goserelin are indicated in the treatment of advanced prostate cancer and more recently these agents have been incorporated as part of neoadjuvant therapy of early-stage prostate cancer.
Aromatase Inhibitors
The aromatase reaction is responsible for the extra –adrenal synthesis of estrogen from androstenedione, which takes place in liver, fat , muscle , skin and breast tissue
Peripheral aromatization is an important source of estrogen in post menopausal women
Aromatase inhibitors decrease the production of estrogen in these women
Eg
Aminoglutethimide , Anastrozole, letrozole , Exemestane

Aminoglutethimide
It was the first aromatase inhibitor to be identified for the treatment of metastatic breast cancer in postmenopausal women
Mechanism of action
It was shown to inhibit both the adrenal synthesis of pregnenolone ( a precursor of estrogen) from cholesterol
It also inhibits the extra-adrenal synthesis of estrone and estradiol.
Aside from its direct effects on adrenal steroid genesis, aminoglutethimide is an inhibitor of an aromatase enzyme that converts the adrenal androgen androstenedione to estrone
This aromatization of an androgenic precursor into an estrogen occurs in body fat.
Aminoglutethimide is normally administered with hydrocortisone to prevent symptoms of adrenal insufficiency.
Hydrocortisone is preferable to dexamethasone because the latter agent accelerates the rate of catabolism of aminoglutethimide.
Uses
Since estrogens promote the growth of breast cancer, estrogen synthesis in adipose tissue can be important in breast cancer growth in postmenopausal women.
Aminoglutethimide is primarily used in the treatment of metastatic breast cancer in women whose tumors express significant levels of estrogen or progesterone receptors.
Dis –advantage
Due to its nonselective properties and unfavorable side effects as well as the need to concomittanity administer hydrocortisone (cortisol) , newer aromatase inhibitors has been developed
Anastrozole , Letrozole & Exemestane

Anastrozole & Letrozole
These are imidazole aromatase inhibitors and are non- steroidal
These are considered to be second – line therapy after tamoxifen for hormone – dependent breast cancer in the united states
They have become first –line drugs in other countries for the treatment of breast cancer in postmenopausal women
They have better action then the aninoglutethimide due to
1.They are more potent by greater than 96 %
2. They are more selective than aminoglutethimide
3. They do not need to be supplemented with hydrocortisone
4. They do not predispose to endometrial cancer
They are orally active and cause almost a total suppression of estrogen synthesis
They are cleared primarily by liver metabolism
Exemestane
It is a steroidal hormonal agent and irreversibly inhibitor of aromatase.
It is orally will- absorbed and widely distributed
Hepatic; metabolism is by the CYP3A4 isozyme , but todate , no interactions have been reported
Because the metabolites are excreted into the urine
The doss of the drug must be adjusted in patients with renal failure
Its major toxicities are nausea, fatigue and loss of hair
This agent is indicated for the treatment of advanced breast cancer in postmenopausal women whose disease has progressed on tamoxifen therapy
X. Miscellaneous
Cisplatin
This is a platinum containing compound, the chemical structure of which is cis- diammine dichloro-platinumMechanism of action
Cisplatin, carboplatin and oxaliplatin are platinum containing compounds . They get converted to the active form in the cell, inhibit DNA synthesis and cause cytotoxicity
A heavy metal alkylator of DNA. Covalently bonds to proteins, RNA and especially DNA, forming DNA cross linking and intra-strand N-7 adduct.
The trans isomer has virtually no anti-tumour activity
Metabolism
It is widely distributed in the body , except for the CNS
Long half – life in plasma (upto 3 days ) may remain bound in tissues for months
Native drug (30%) and metabolites excreted in urine , biliary excretion accounts for less than 10% of the total drug excretion
Administration
The principles of cisplatin administration are as follows
1. Monitoring of serum cratinine, electrolytes , magnesium and calcium levels daily
during therapy
2. Anti- emetics such as ondansetron and dexamethasone should be administered
Prophylactically
3. Hydration and diuresis are required when 40mg /m2 or more of cisplatin is given to
maintain a urine output of 100 -150 ml /hour before administration of the drug
4. Furosemide is given to prevent fluid overload
5. Mannitol Is given if urine output is insufficient
6. Intravenous fluids are supplemented with KCL and Mg SO4
Preparation
Inj - 10 mg /20 ml , 50 mg/100 ml
Dosage
It is available as 10 and 50 mg vials
40 – 120 mg/m2 or more intravenously every 3-4 weeks or 20 – 40 mg/m2 daily for 3 – 5 days every 3 – 4 weeks
Adverse effects
It causes ototoxicity, nephrotoxicity, peripheral neuropathy, nausea, vomiting , and anemia. It is relatively less toxic to bone marrow
Dose modification
Renal function must return to normal before cisplatin can be given . One may avoid using cisplatin when the cratinine clearance is less than 40 ml/ min .
It is relatively contraindicated in patient with documented hearing impairment
Therapeutic uses
Cisplatin is effective in advanced colorectal cancer and in other cancers like ovarian and cervical cancers
Carboplatin
It is a second-generation platinum analog that exerts its cytotoxic effects exactly as cisplatin and has activity against the same spectrum of solid tumors.
Its main dose-limiting toxicity is myelosuppression, and it has significantly less renal toxicity and gastrointestinal toxicity than cisplatin.
Moreover, vigorous intravenously hydration is not required.
As a result, carboplatin is now being used in place of cisplatin in combination chemotherapy.
Oxaliplatin
It is a third generation diaminocyclohexane platinum analog. Its mechanism of action is identical to that of cisplatin and carboplatin.
However, it is not cross-resistant to cancer cells that are resistant to cisplatin or carboplatin on the basis of mismatch repair defects.
This agent was recently approved for use as second-line therapy in metastatic colorectal cancer following treatment with the combination of fluorouracil-leucovorin and irinotecan, and it is now widely used as first line therapy of this disease as well.
Neurotoxicity is dose-limiting and characterized by a peripheral sensory neuropathy, often triggered or worsened upon exposure to cold.
Imatinib

Mechanism of action
It occupies the ATP biding site of eh BCR- ABL protein in the myeloma cells ( in CNL patients) and other related tyrosine kinases and thus results in subsequent inhibition of substrate phosphorylation
It is a protein and selective inhibitor resulting in inhibition of tumourigenicity of BCR – ABL positive cells and also leads to their apoptosis
It also inhibits other receptor tyrosine kinases for platelet derived growth factor receptor (PDGFR)
Metabolism
It is eliminated mainly in faeces. The half – life is around 18 hours
Adverse effects
Nausea, vomiting , diarrhea , pulmonary oedema& myelosuppression
Pharmacokinetics
Imatinib is administered orally and is well absorbed; it is highly protein-bound in plasma.
The drug is metabolized in the liver, and elimination of metabolites occurs mainly in feces via biliary excretion.
Administration
It is supplied as 100 mg capsules and should be taken with food and a large glass of water
The dose modifications required in patients with liver dysfunction
Dosage
400 mg/day orally for Chronic myelogenous leukemia in chronic phase and in Gastrointestinal stromal tumours, 600 – 800 mg/ day orally in accelerated phase of CML
Preparation
Imatinib – Cap -100 mg
Uses
It is used in the treatment of chronic myelogenous leukaemia(CML) and gastrointestinal stromal tumours(GIST)
This agent is approved for use as first-line therapy in chronic phase CML, in blast crisis, and as second-line therapy for chronic phase CML that has progressed on prior intestinal stromal tumours.
Imatinib is effective also for treatment of gastrointestinal stromal tumors expressing the
Gefitinib

Mechanism
It inhibits the enzyme tyrosine kinase associated with transmembrane cell surface receptors , including the epidermal growth factor receptor (EGFR)
Metabolism
It undergoes extensive hepatic metabolism and is excreted predominantly in faeces
Administration
It is supplied as 250 mg tablets. Dose modification s required in patients with liver dysfunction
Dosage
250 mg orally
Preparation
Gefitinib - Tab - 250 mg
Adverse effects
Diarrhea, nausea, vomiting , mild rash , anorexia , peripheral oedema and corneal irritation
Uses
Advanced non small cell lung cancer and taxane – based therapies
Hydroxyurea

Mechanism of action
Hydroxyurea (HONHCONH2) is an analog of urea whose mechanism of action involves the inhibition of DNA synthesis in the S phase by inhibiting the enzyme ribonucleotide reductase, resulting in depletion of deoxynucleoside triphosphate pools.
Pharmacokinets
The drug is administered orally and has nearly 100% oral bioavailability.
Metabolism in liver and excreted by kidney
It is mainly used in chronic myelogenous leukemia and treatment of the blast crisis of acute myeloid leukemia.
Adverse effects
Myelosuppression is the dose-limiting toxicity, but nausea and vomiting, mucositis and diarrhea, headache and increased lethargy, and a skin rash.
Uses
It is also effective as an adjunct with radiation therapy for head and neck cancer and in treating essential thrombocytosis and polycythemia vera.
